 Hydrogen serves as one of the key building blocks in many chemical industries (e.g.ammonia synthesis, oil refinery) and has been advocated as a promising alternative to fossil fuels with zero emission. Currently, nearly 95% of the global hydrogen production is derived from the steam reforming process, which generates a considerable amount of greenhouse gas emissions. In fact, the corresponding outlet of CO2 is around 10 times that of H2production. In the past few decades, electrocatalytic water splitting (also known as water electrolysis) emerges as an attractive technology for sustainable hydrogen production with zero greenhouse gas emission. Further more, it also provides the potential synergy with renewable energy resources that are intermittently available (e.g.solar, wind). However, hydrogen production from water electrolysis is still 3-10 times more expensive than that from steam reforming, impeding its industrial-scale implementation.
Hydrogen serves as one of the key building blocks in many chemical industries (e.g.ammonia synthesis, oil refinery) and has been advocated as a promising alternative to fossil fuels with zero emission. Currently, nearly 95% of the global hydrogen production is derived from the steam reforming process, which generates a considerable amount of greenhouse gas emissions. In fact, the corresponding outlet of CO2 is around 10 times that of H2production. In the past few decades, electrocatalytic water splitting (also known as water electrolysis) emerges as an attractive technology for sustainable hydrogen production with zero greenhouse gas emission. Further more, it also provides the potential synergy with renewable energy resources that are intermittently available (e.g.solar, wind). However, hydrogen production from water electrolysis is still 3-10 times more expensive than that from steam reforming, impeding its industrial-scale implementation.
Electrolysis of water comprises two half-reactions, the hydrogen evolution (HER) at the cathode and the OER at the anode.
HER: 4H++4e- → 2H2(g)
OER: 2H2O(l) → O2(g)+4H++4e-
Although the thermodynamic potential to drive water electrolysis is only 1.23 V at standard conditions, excess energy in the form of overpotential is always required to split water at a realistic rate. A typical polymer-electrolyte-membrane (PEM) electrolyzer with Pt as HER catalyst and Ir/Ru oxide mixture as OER catalyst, exhibits a total overpotential between 600-1000 mV under the operational current density of 0.6 to 2A cm-2. The use of high cost and scarce noble metal catalysts, and low energy efficiency are two dominant factors in determining the cost of electrolytic hydrogen. Consequently, developing efficient water splitting electrocatalysts with non-precious metals becomes a significant theme to make sustainable hydrogen production affordable. Over the past decade, earth-abundant materials including chalcogenides, phosphides, and nitrides have been explored as both HER and OER catalysts. Among them, transition metal phosphides(TMPs) have been outstanding with excellent conductivity, decent corrosion resistance, and the potential as HER/OER bifunctional catalysts. However, substantial challenges remain to be tackled before the broad application of TMPs for water electrolysis, including but not limited to:
(a) Engineering the TMP materials for better HER performance (i.e.lower overpotential).
(b) Understanding the OER chemistry on TMPs.
To address the above challenges, the following research program consisting of two projects specifically targeting OER and HER will be conducted t. Thorough theoretical investigations by means of density functional theory (DFT) will be performed to design highly efficient TMP-based water electrolysis catalysts and develop an atomistic understanding of the structure-property correlations for TMPs materials
Project (a): Computational design of transition metal@ phosphide NPs for the HER
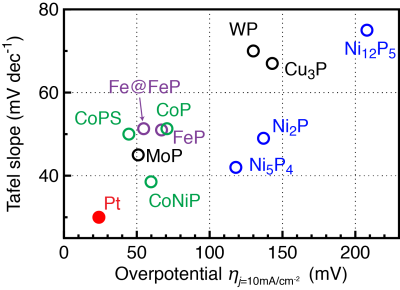
Immense experimental efforts have been devoted to improving the HER performance of TMP catalysts. Several strategies, including the development of ternary complex compounds, phase controlled synthesis, partial phosphidation showed effective reductions of the HER overpotential. However, few of them surpassed the performance of the benchmark Pt catalyst, as summarized in Figure 1. On the other hand, the insight from theory is still limited, which requires rigorous studies.
Inspired by the partial phosphidation preparation of Fe@FeP NPs, we propose a new design approach using transition metal@phosphide (TM@TMP) core-shell architecture to search for Pt alternatives. The intent underlying this design is to use the transition metal core to tailor the catalytic properties of TMP shell. My previous work on core-shell metallic NPs highlighted that strain and charge transfer from the core necessarily modified the surface reactivity. We hypothesize that the same tuning mechanism holds for TMP materials as well, which can be first tested by DFT simulations and further validated through potential collaboration with experimental groups. In principle, the TM@TMP NPs can be achieved through partial phosphidation of TM@TM or TM@TMO NPs, where the core and shell metals are not necessarily the same.
As a starting point, our effort will center on FeP-based materials, since Fe is the most earth-abundant transition metal. FeP crystalizes in an orthorhombic MnP-type structure (Pnma space group). X-ray diffraction (XRD) patterns in previous experimental studies on FeP NPs showed that a number of surface orientations, such as (002), (011), (200), (103) and (111) could be stabilized on different supports. We will first identify the most relevant surface orientation for the HER by computing the surface energies as well as the theoretical HER overpotentials in terms of ∆GH. Subsequently, the metal-phosphide interface will be constructed using Coincidence Site Lattice (CSL) method.
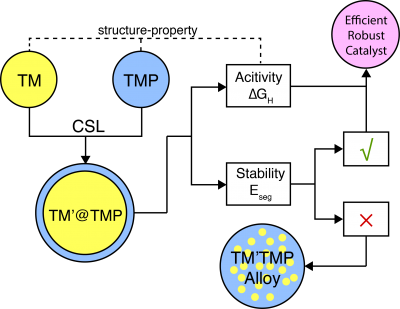
Following the same procedure, we can efficiently build computational models for TM@FeP NPs with different core metals and investigate the corresponding HER performance. Subsequent electronic structure analysis, including Bader charges, the density of states, and band structures will be performed to correlate the core metal and the HER activity on the FeP shell. The HER performance of pure FeP with a range of biaxial-strain in the XY plane will be evaluated as a reference to further disentangle the strain effect and charge transfer. In the case that the core metal is not Fe, we will also assess the thermodynamic stability of TM@FeP NPs by computing the core/shell segregation energy, which is defined as the energy required to swap a surface Fe atom with a core metal atom. If the segregation is favored, the HER activity of the segregated heteroatom and its nearby sites will be investigated, which leads to the study of the ternary phosphide solid solution.
With the accomplishment of the study on FeP-shelled NPs, a general theoretical framework (Figure 2) will be developed to study and design other TM@TMP catalysts for the HER. The proposed research will provide a solid footing for the development of TMP-based HER catalysts and shed light on the application of TMPs for other energy-related problems (such as selective hydrogenation, hydrodenitrogenation, and hydrodesulfurization).
Project (b): Fundamental understanding of the OER chemistry on TMPs
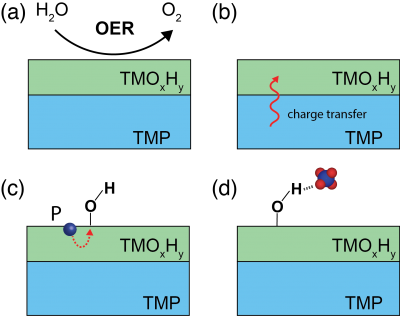
Recently, there has also been an increasing interest in TMPs for the application in the OER. Several TMPs, such as CoP, NixP, and FeP have shown decent OER activities that are even comparable to the state-of-the-art IrO2catalysts. Rigorous postcatalysis characterizations and analysis revealed that the surface of TMP catalysts was irreversibly oxidized and transformed to the corresponding oxide or oxyhydroxide form (TMOxHy) under practical OER conditions, as shown in Figure 3(a). In other words, TMPs are most likely to be the precursor to its real OER active form. Interestingly, many of these TMP-transformed catalysts exhibit superior activity to their oxide or oxyhydroxide counterparts. All this leads to an open question for the OER on TMP catalyst: “whether phosphorus in any form plays any role in the enhanced OER activity”, where insight from theory is highly demanded.
To answer this question, We propose a thorough theoretical investigation of the TMP/TMOxHyheterostructure, where TMP is completely covered by one or multiple layer(s) of corresponding (oxy)hydroxides. The proposed hetero-structure provides a model system for the in-depth understanding of the OER chemistry on TMP catalysts. Such a heterostructured model will be built following using the same approach as introduced above for the metal/phosphide interface. We will first study the FeP system since some of the structures in the project 2(a) can be reused here. Virtual experiments with theoretical simulations will be performed on this model system to test the following three hypotheses, as illustrated in Figure 3(b-d):
-
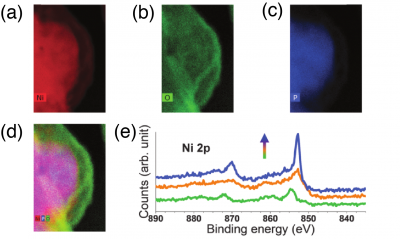
Figure 4. Corresponding EDX maps of (a) Nickel, (b) Oxygen, (c) Phosphorus, (d) Combined elemental mapping of Ni, O, and P. (e) High-resolution depth-profiling XPS spectra of the Ni 2p area. As the profiling depth increases (arrow direction). Courtesy of Hu et. al. Charge transfer between TMP substrate and surface TMOxHy improves the surface OER activity. In some of these studies, TMP remained in the core and only the surface was oxidized, as shown in Figure 4(a-d). Thus, it is possible that the observed enhancement is due to a charge transfer from the underlying TMP core to the catalytically active TMOxHy surface, which changes oxidation states of the surface TM We will compare the OER activity of TMOxHy layer with and without the underlying TMP core to examine the support effect. Detailed electronic structure analysis on surface TM oxidation states will also be conducted to quantify the charge transfer from the TMP/TMOxHyinteraction.
- Traces of P that incorporated in the TMOxHy surface enhances the OER activity. As shown in Figure 4(e), during the oxidation of Ni2P, surface P was mostly oxidized to phosphate. However, XPS detectable amount of P remained on the surface (the green profile). These residual P atoms may incorporate into the TMOxHysurface and locally influence the activity of neighboring sites. A Pourbaix diagram based on comprehensive DFT calculations will be constructed to determine whether and where the incorporation of P will occur. Eventually, using the most relevant structure from the Pourbaix analysis, the synergistic effect of P incorporation on the OER performance will be evaluated.
- Phosphate ion acts as a catalytic promoter for the OER. Another possible form of residual P is surface-bound phosphate group. A recent study in the Brock group suggested that the activity decay may couple with the surface-bound phosphate leaching. Phosphate can serve as either a catalytic promoter improving the performance of TMOxHy surface or an effective buffer to accelerate the proton-coupled electron transfer (PCET) processes during the OER. We will assess its role as a promoter first by computing the reaction free energy diagram with and without the presence of phosphate. Afterward, kinetics of deprotonation steps by the water molecule and phosphate ion will be studied to shed light on the involvement of phosphate in PCET.